Case 3: Benchmark and Method validation#
In this example, we show how to compare the results from implicit and explicit H-bond methods.
Import the required packages#
import os
import prolif as plf
from prolif import sdf_supplier
from prolif.io.protein_helper import ProteinHelper
from prolif.molecule import Molecule
from prolif.plotting.complex3d import Complex3D
from rdkit import Chem
/home/yuyang/Project_local/GSoC2025_Hbond_PM/.venv/lib/python3.11/site-packages/MDAnalysis/topology/tables.py:52: DeprecationWarning: Deprecated in version 2.8.0
MDAnalysis.topology.tables has been moved to MDAnalysis.guesser.tables. This import point will be removed in MDAnalysis version 3.0.0
warnings.warn(wmsg, category=DeprecationWarning)
# custom functions for comparison and plotting
from validation.utils.metrics import (
confusion_matrix,
get_interactions,
plot_confusion_matrix,
tanimoto_coefficient_by_confusion_matrix,
)
We also use the protein helper function to read the files.
protein_helper = ProteinHelper(
templates=[
{
"MSE": {"SMILES": "C[Se]CC[CH](N)C=O"},
}
]
)
The test case is from PLINDER: 5da9__1__1.A_1.B__1.E_1.F. We have prepared the protonated file in the repository.
test_case_dir = "./test_data/5da9__1__1.A_1.B__1.E_1.F"
Explicit H-bond method#
First, we read the molecule from the path.
protein_mol = Molecule.from_rdkit(
Chem.MolFromPDBFile(
f"{test_case_dir}/receptor_protonated.pdb",
sanitize=False,
removeHs=False,
proximityBonding=True,
)
)
protein_mol = protein_helper.standardize_protein(protein_mol)
Then, we check the length of the residues.
protein_mol.residues.__len__()
834
We also check with the residues to know whether they are fixed with our protein helper as expected.
plf.display_residues(protein_mol, slice(0, 10), sanitize=False)

We next load all ligands in the directory.
ligands = []
for ligand_sdf in os.listdir(test_case_dir): # noqa: PTH208
if ligand_sdf.endswith("_protonated.sdf"):
ligands.extend(sdf_supplier(f"{test_case_dir}/{ligand_sdf}"))
There are two ligands in our directory.
ligands
[<prolif.molecule.Molecule with 1 residues and 47 atoms at 0x7fb4128bb3d0>,
<prolif.molecule.Molecule with 1 residues and 1 atoms at 0x7fb4129c3510>]
plf.display_residues(ligands[0])

We currently focus on the cation/anion one.
ligand = ligands[0]
Then, we calculate the H-bond interactions (using explicit hydrogens).
fp = plf.Fingerprint(["HBDonor", "HBAcceptor"], count=True)
fp.run_from_iterable([ligand], protein_mol, progress=False)
df = fp.to_dataframe().T
Here is the result.
df
| Frame | 0 | ||
|---|---|---|---|
| ligand | protein | interaction | |
| UNL1 | ARG13.A | HBAcceptor | 1 |
| ASN36.A | HBAcceptor | 1 | |
| GLY37.A | HBAcceptor | 1 | |
| GLY39.A | HBAcceptor | 1 | |
| LYS40.A | HBAcceptor | 1 | |
| THR41.A | HBAcceptor | 1 | |
| THR42.A | HBAcceptor | 1 | |
| ALA64.A | HBDonor | 1 | |
| ASP68.A | HBAcceptor | 1 | |
| GLN159.A | HBAcceptor | 1 | |
| ARG431.A | HBAcceptor | 1 | |
| GLY344.B | HBAcceptor | 1 | |
| GLN345.B | HBAcceptor | 1 |
2D visualization#
We visualize the result into an 2D plot.
view = fp.plot_lignetwork(ligand, kind="frame", frame=0, display_all=False)
view
3D visualization#
The result can also be visualized in 3D plot.
view = fp.plot_3d(ligand, protein_mol, frame=0, display_all=True)
view
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
<prolif.plotting.complex3d.Complex3D at 0x7fb4128211d0>
Implicit H-bond method#
To have a better comparison, we simply remove the hydrogens from the object instead of reading a new file without explicit hydrogens.
protein_mol_i = protein_helper.standardize_protein(
Molecule.from_rdkit(Chem.RemoveAllHs(protein_mol))
)
Again, we check the residues after our protein helper.
plf.display_residues(protein_mol_i, slice(0, 10), sanitize=False)

Same for the ligand, we remove the hydrogens.
ligand_i = Molecule.from_rdkit(Chem.RemoveAllHs(ligand))
We then can recalculate the H-bond interations (but with implicit hydrogens).
fp_i = plf.Fingerprint(
["ImplicitHBDonor", "ImplicitHBAcceptor"],
count=True,
)
fp_i.run_from_iterable([ligand_i], protein_mol_i, progress=False)
df_i = fp_i.to_dataframe().T
Here is the result.
df_i
| Frame | 0 | ||
|---|---|---|---|
| ligand | protein | interaction | |
| UNL1 | ARG13.A | ImplicitHBAcceptor | 1 |
| ASN36.A | ImplicitHBAcceptor | 1 | |
| GLY37.A | ImplicitHBDonor | 1 | |
| ImplicitHBAcceptor | 1 | ||
| SER38.A | ImplicitHBDonor | 1 | |
| ImplicitHBAcceptor | 2 | ||
| GLY39.A | ImplicitHBAcceptor | 1 | |
| LYS40.A | ImplicitHBDonor | 1 | |
| ImplicitHBAcceptor | 2 | ||
| THR41.A | ImplicitHBAcceptor | 3 | |
| THR42.A | ImplicitHBAcceptor | 2 | |
| ALA64.A | ImplicitHBDonor | 1 | |
| ASP68.A | ImplicitHBAcceptor | 1 | |
| SER342.B | ImplicitHBDonor | 1 | |
| ImplicitHBAcceptor | 1 | ||
| GLY344.B | ImplicitHBAcceptor | 1 | |
| GLN345.B | ImplicitHBAcceptor | 1 |
2D visualization#
view = fp_i.plot_lignetwork(ligand_i, kind="frame", display_all=False)
view
3D visualization#
view = fp_i.plot_3d(ligand_i, protein_mol_i, frame=0, display_all=True)
view
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
<prolif.plotting.complex3d.Complex3D at 0x7fb412a73150>
Comparison between explicit and implicit H-bond methods#
For the 3D visualization, we can plot the results side by side using compare function. The left panel shows the H-bond interations from explicit hydrogens and the right panel is those from implicit hydrogens.
# create Complex3D objects (left: explicit)
comp3D = Complex3D.from_fingerprint(fp, ligand, protein_mol, frame=0)
# (right: implicit)
other_comp3D = Complex3D.from_fingerprint(fp_i, ligand_i, protein_mol_i, frame=0)
# compare the two Complex3D objects
view = comp3D.compare(other_comp3D, display_all=True)
view
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
<prolif.plotting.complex3d.Complex3D at 0x7fb4128b0a90>
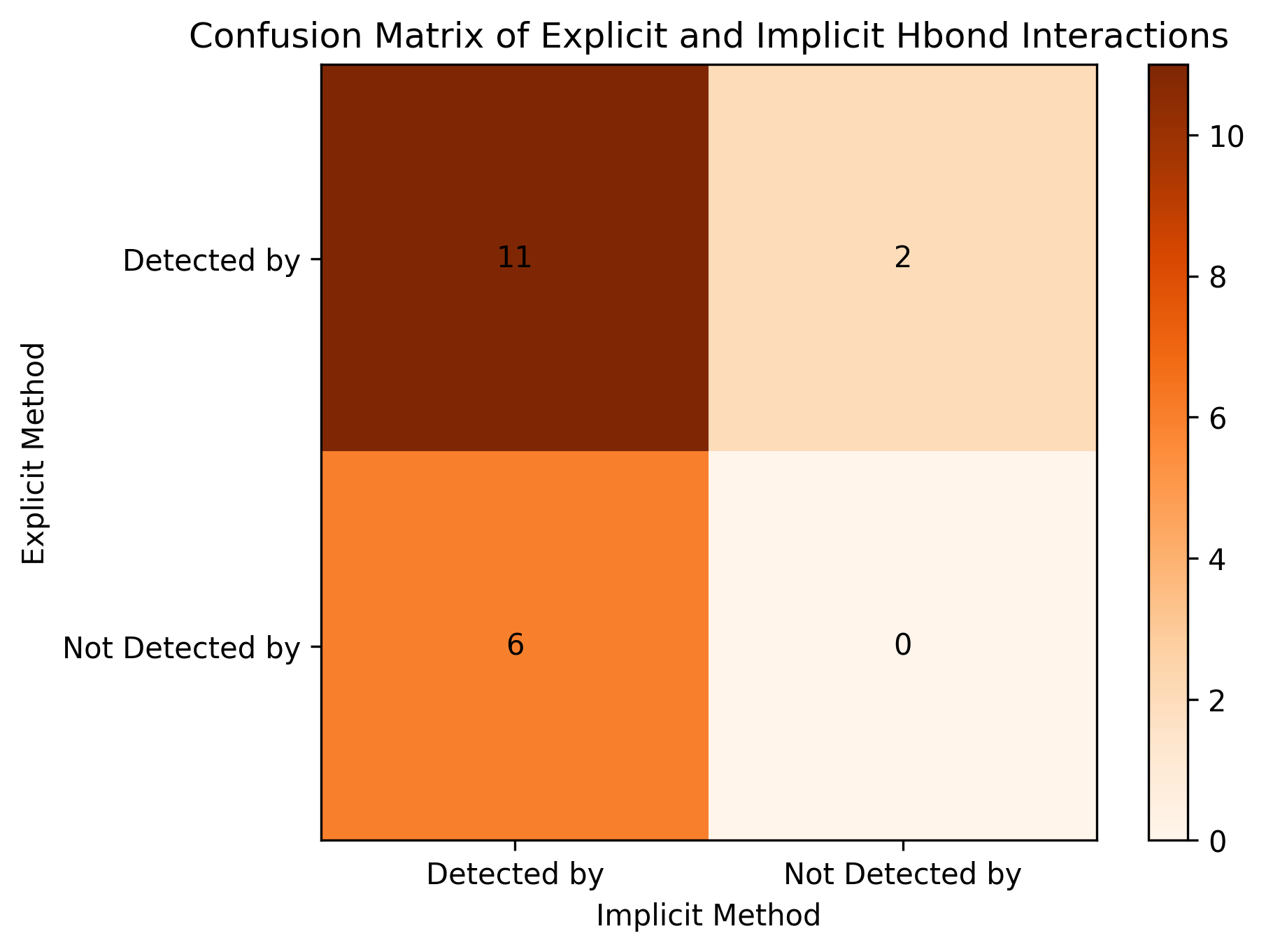
For the interation pairs, we can covert the result into two sets explicit_set and implicit_set. Then, we can know how many interactions detected by the explicit H-bond method overlap with those detected by the implicit H-bond method.
explicit_set = get_interactions(df)
implicit_set = get_interactions(df_i)
matrix = confusion_matrix(explicit_set, implicit_set)
tm_coef = tanimoto_coefficient_by_confusion_matrix(matrix)
The Tanimoto coefficient (also called intersection over union) is a measure used to evaluate performance. A higher score indicates better performance.
print("Tanimoto coefficient:", tm_coef) # noqa: T201
fig, ax = plot_confusion_matrix(matrix)
fig.show()
Tanimoto coefficient: 0.5789473684210527